Discovery-based Quantitative MS methods
Discovery-based Quantitative MS methods,(“shotgun” proteomics), aim to identify as many proteins in a sample as possible and can be completed on a wide range of samples with minimal method development.
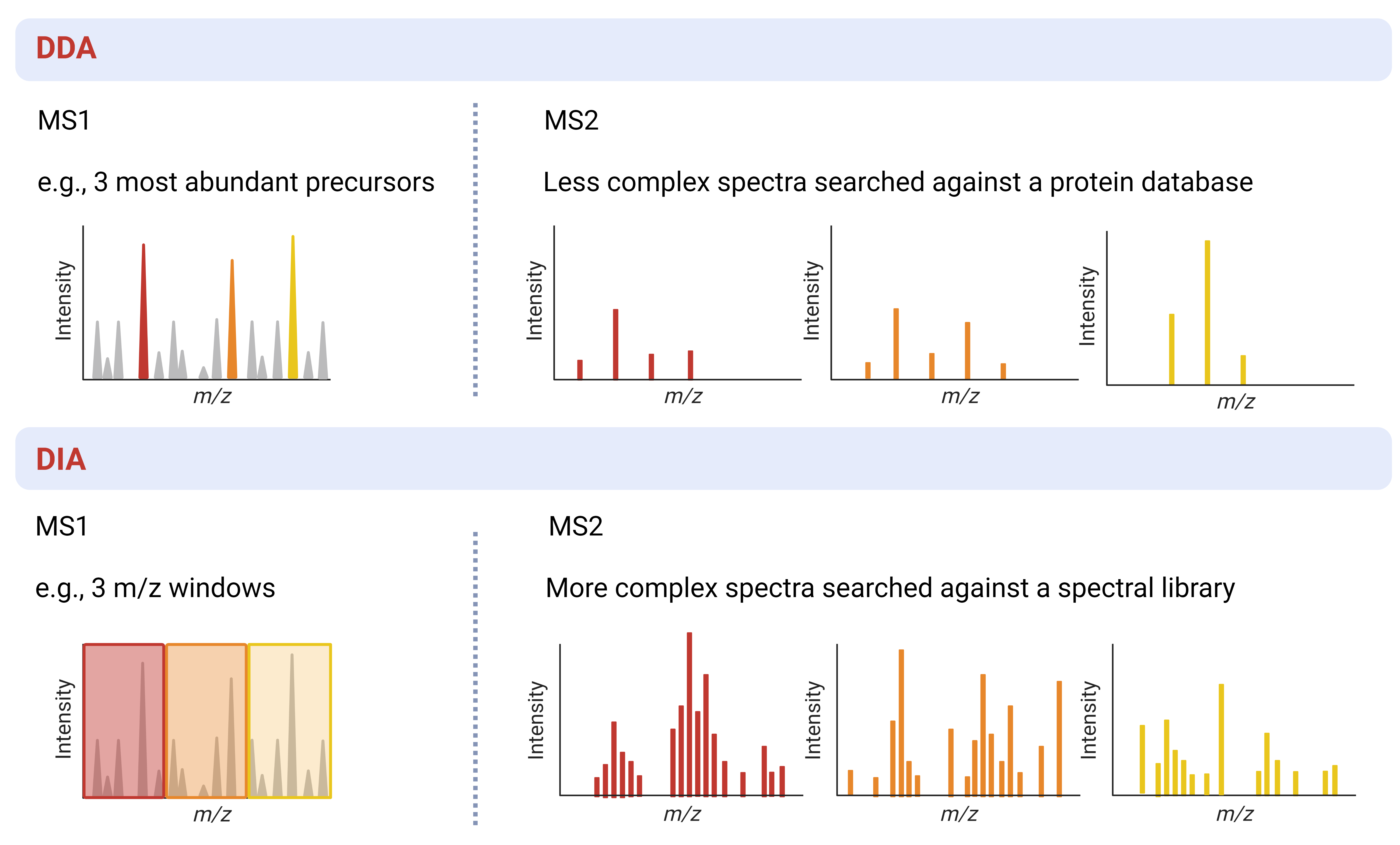
- DDA (data-dependent acquisition): In Data-dependent acquisition (DDA) mode a MS instrument will complete a full scan of all peptide (precursor) ions during MS1 and then select the most abundant ions for fragmentation and MS2
- DIA (data-independent acquisition): In Data-independent acquisition (DIA) mode a MS instrument scans pre-determined, often sequential, m/z ranges and then fragments all the ions within each range for MS2 measurement
| DDA | DIA | |
|---|---|---|
| Suitable for small proteomes* | Yes | Yes |
| Suitable for large proteomes* | No | Yes |
| Dynamic range | Moderate | High |
| Complexity of spectra | Low | High |
| Search requirements | Protein database | Spectral library |
| Instrument method optimization | Low | Hig |
Note (*) Most prokaryote proteomes can be considered a “small” proteome, vs. larger eukaryotic or mixed (e.g., infection model) proteomes
Discovery-based methods can be used to identify thousands of proteins in a single sample, and for most research questions label-free DDA is a straight-forward option that has relatively low reagents costs, and requires minimal sample preparation and instrument method optimization.